-
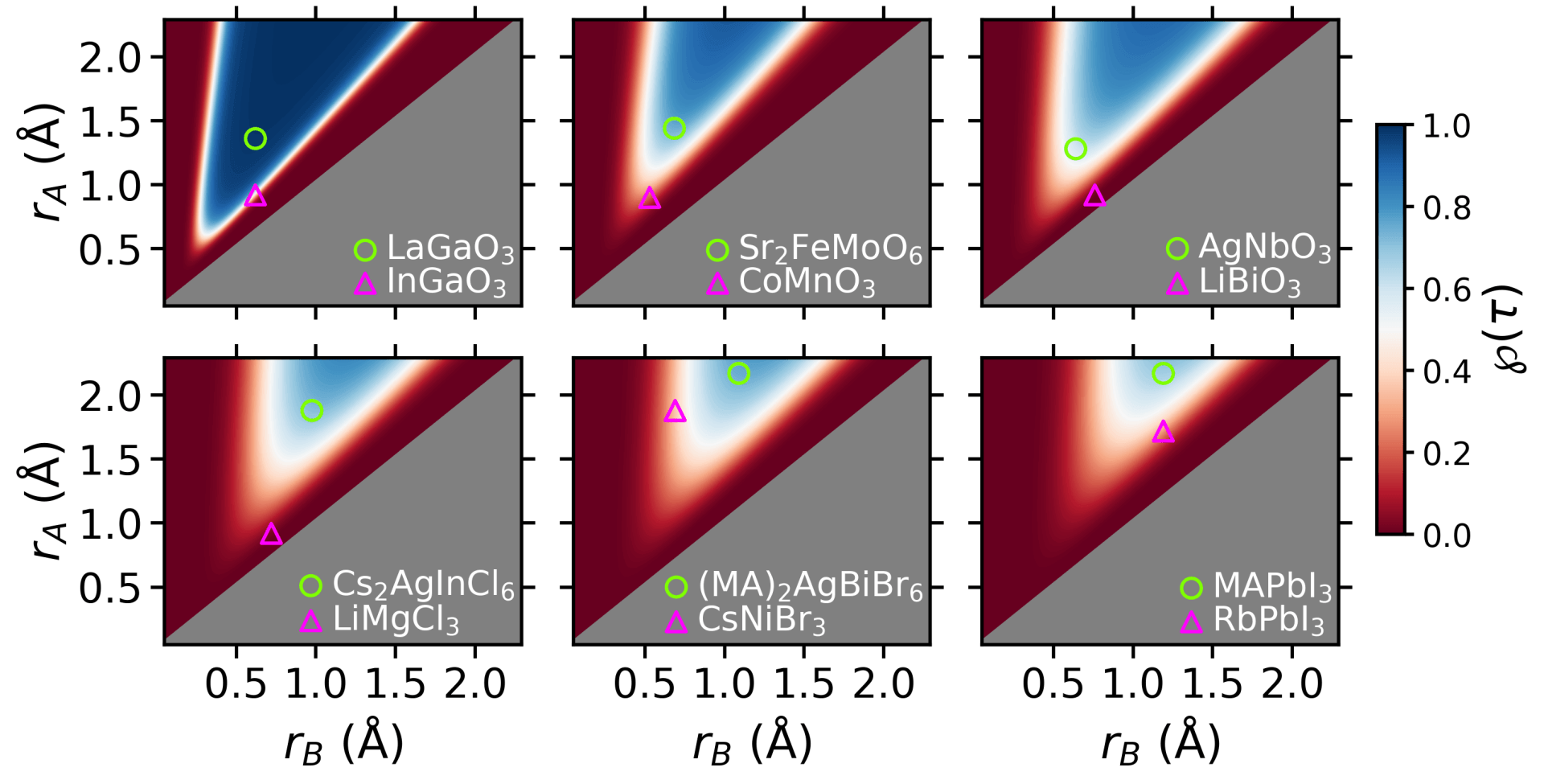
New Tolerance Factor to Predict the Stability of Perovskite Oxides and Halides
Button -

Noncovalent intermolecular interactions in organic electronic materials: implications for the molecular packing vs electronic properties of acenes
Button -

Machine learning for heterogeneous catalyst design and discovery
Button -
Slide title
Write your caption hereButton 
Slide title
Write your caption hereButton
Slide title
Write your caption hereButton
Welcome to
Christopher Sutton's
research page on
computational chemistry and materials science
-
New Tolerance Factor to Predict the Stability of Perovskite Oxides and Halides
Button -
Noncovalent intermolecular interactions in organic electronic materials: implications for the molecular packing vs electronic properties of acenes
Button -
Machine learning for heterogeneous catalyst design and discovery
Button -
Slide title
Write your caption hereButton -
Slide title
Write your caption hereButton -
Slide title
Write your caption hereButton
Machine
Learning Methods
Learning Methods
Computational Materials Discovery
Machine Learning Methods
Computational Materials Discovery
New Tolerance Factor to Predict the Stability of Perovskite Oxides and Halides
- …read more
… (or tolerance factor) τ, that correctly classifies 92% of compounds as perovskite or nonperovskite for an experimental dataset containing 576 ABX3 materials (X = O2-, F-, Cl-, Br-, I-). In comparison, the widely used Goldschmidt tolerance factor, t, achieves a maximum accuracy of only 74% for the same set of materials. Currently our work is focused on investigating our predictions with DFT on a double perovskites with formula A2BB’X6.
New Tolerance Factor to Predict the Stability of Perovskite Oxides and Halides
ButtonNew Tolerance Factor to Predict the Stability of Perovskite Oxides and Halides
- …read more
… (or tolerance factor) τ, that correctly classifies 92% of compounds as perovskite or nonperovskite for an experimental dataset containing 576 ABX3 materials (X = O2-, F-, Cl-, Br-, I-). In comparison, the widely used Goldschmidt tolerance factor, t, achieves a maximum accuracy of only 74% for the same set of materials. Currently our work is focused on investigating our predictions with DFT on a double perovskites with formula A2BB’X6.
Noncovalent intermolecular interactions in organic electronic materials: implications for the molecular packing vs electronic properties of acenes
- …read more
… Here, we provide an overview of the theoretical underpinnings of noncovalent intermolecular interactions and briefly discuss the computational chemistry approaches used to understand the magnitude of these interactions. These methodologies are then exploited to illustrate how noncovalent intermolecular interactions impact important electronic propertiessuch as the electronic coupling between adjacent molecules, a key parameter for charge-carrier transport through a comparison between the prototype organic semiconductor pentacene with a series of N-substituted heteropentacenes.
Machine Learning for Heterogeneous Catalyst Design and Discovery
Noncovalent intermolecular interactions in organic electronic materials: implications for the molecular packing vs electronic properties of acenes
- …read more
… Here, we provide an overview of the theoretical underpinnings of noncovalent intermolecular interactions and briefly discuss the computational chemistry approaches used to understand the magnitude of these interactions. These methodologies are then exploited to illustrate how noncovalent intermolecular interactions impact important electronic propertiessuch as the electronic coupling between adjacent molecules, a key parameter for charge-carrier transport through a comparison between the prototype organic semiconductor pentacene with a series of N-substituted heteropentacenes.
Machine Learning for Heterogeneous Catalyst Design and Discovery
